Mantle cell lymphoma (MCL) is a mature B-cell neoplasm, which represents 2-3% of cases of non-Hodgkin lymphoma in the US. The IgH/Cyclin D1 translocation is characteristic of this lymphoma, and can be confirmed by FISH testing in almost all cases. Clincially, patients often present with widespread disease. In addition to adenopathy, patient often have involvement of blood (20-40%) and other organ sites (gastrointestinal tract, liver spleen, bone marrow).
Morphology
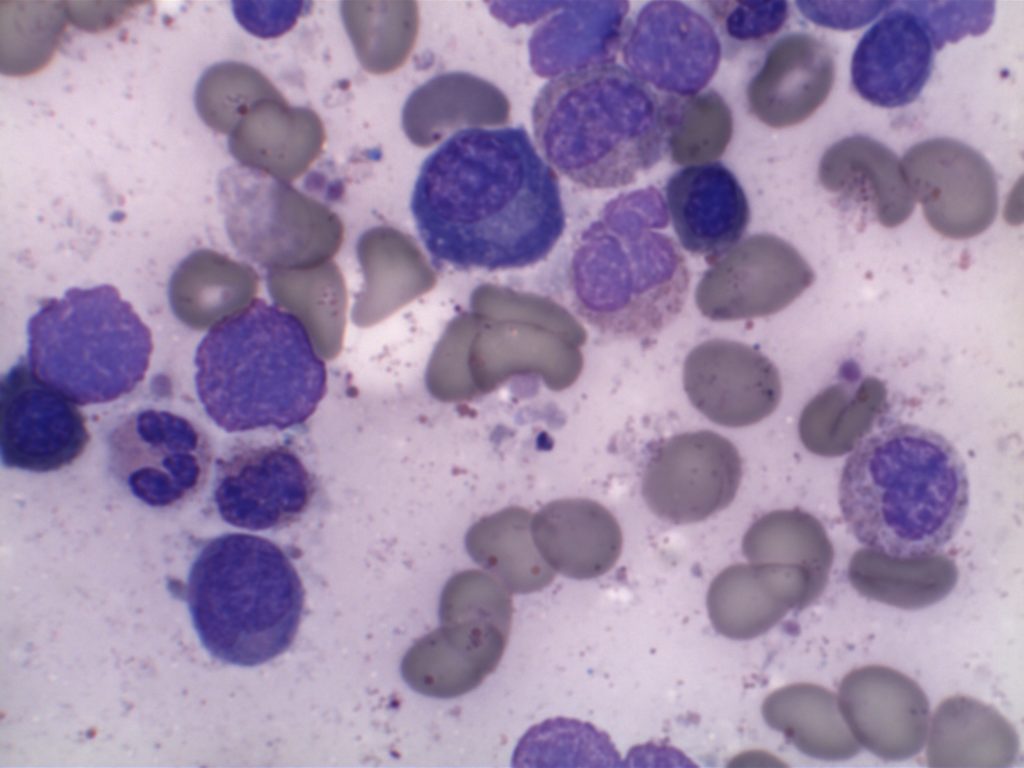
MCL is typically characterized by a small to intermediate sized lymphocytes with an irregular nuclear membrane (CLL/SLL tends to have a smoother nuclear membrane and follicular lymphoma has cleaved cells). A subset of cases have a larger size and increased mitotic rate and can be confused with acute lymphoblastic lymphoma, and are referred to as the “blastoid” variant of MCL.
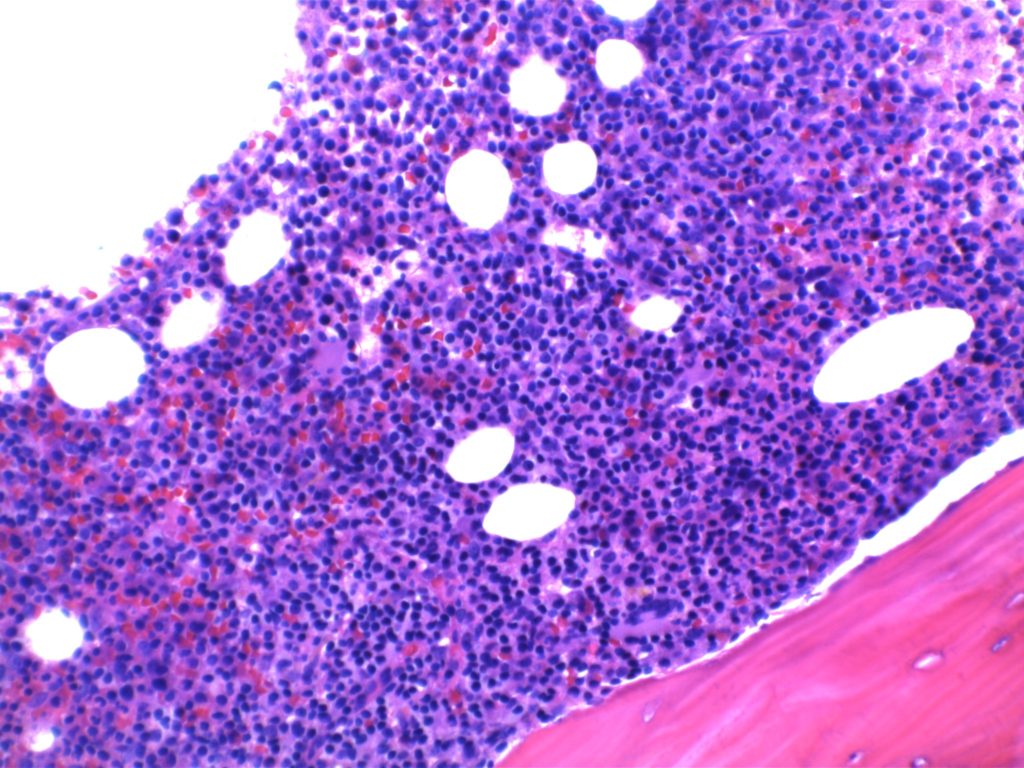
2008 WHO Classification identifies multiple morphologic variant including: blastoid variant (resembles ALL with increased mitoses), pleomorphic (also aggressive), small cell variant (resembles CLL/SLL), and marginal zone-like morphology. Architectural pattern can have a diffuse pattern (often with scattered histiocytes within the infiltrate) or nodular (resembling follicular lymphoma). Minimal lymph node involvement can show expansion of the mantle zones with relative intact lymph node architecture. Bone marrow involvement can have a varied appearance, but can mimic follicular lymphoma with paratrabecular aggregates.
2016 WHO Classification Revision
Two subtypes of mantle cell lymphoma are now recognized, which center around the mutation status of IgHV.
- IgHV unmutated/minimally mutated (mostly SOX11+) – classical disease that is aggressive, typically involves lymph nodes and other extra nodal sites.
- IgHV mutated (SOX11 negative) – associated with indolent non-nodal disease with peripheral blood and bone marrow involvement. Some of these cases may have been difficult to separate from CLL/SLL in the past.
Half of cyclin D1 negative cases show a CCND2 rearrangement.
Molecular Characteristics
- FISH + for t(11;14)
- Cytogenetics + t(11;14) ~70% of cases
- 50% of Cyclin D1 negative cases have CCND2 rearrangements
Immunophenotypic Expression Pattern
|
Marker
|
Comment
|
|
|
Negative
|
|
|
Positive (93-95%). Some data indicates up to 12% of MCL cases may be negative for CD5.
|
|
|
Negative. Up to 8% of cases may express CD10 (expression will usually be <30%).
|
|
|
Positive
|
|
|
Positive
|
|
|
Negative. 21% may express CD23.
|
|
|
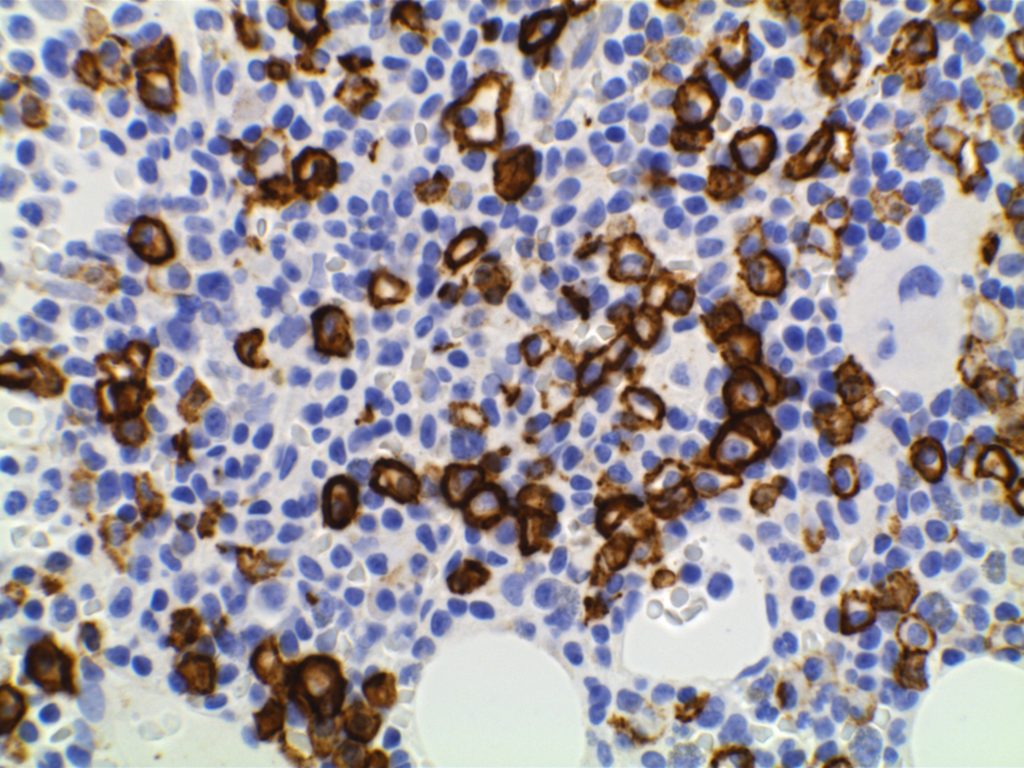
Positive. Nuclear expression. The rabbit monoclonal antibody clone SP4 appears to have the highest sensitivity and stain intensity. Sensitivity ~95%.
|
|
|
Positive
|
|
|
Negative (~12% of cases may have expression)
|
|
|
Usually negative (35% may be positive, of these 2/3rds will also be bcl-6+)
|
|
|
Highlights the residual FDC meshwork.
|
|
|
Inverse relationship between quantitative Ki-67 index and prognosis. Ki-67 >40% is an adverse prognostic factor.
|
|
|
Expressed in classic form and lack of expression is associated with more indolent variant of MCL. |
Important caveats
Cyclin D1 expression is not entirely specific for mantle cell lymphoma. Some expression can be seen in a zonal pattern in CLL/SLL in proliferation centers. Other entities such as plasma cell myeloma (up to 50%) and hairy cell leukemia can also express cyclin D1. CD10 (8%), Bcl-6, and CD23 (21%) has been reported to be expressed in a small number of cases. Loss of CD5 (12%) has been noted in some cases. Most of these studies were by flow cytometry.
CD5 expression on a B-cell lymphoma should practically result in testing for exclusion of a t(11;14) IgH/Cyclin D1 gene fusion. There are other B-cell lymphomas, which may also have CD5 expression (e.g. DLBCL, CLL/SLL, etc.) that are not MCL and have separate prognostic characteristics because of/or separate from CD5 expression.
Rarely, plasmacytic differentiation may be identified. Classic MCL is derived form naive B-cells, which tends to not to progress to plasmacytic differentiation, compared to post germinal center derived B-cell lesions, which more commonly have plasmacytic differentiation.
Ki-67 staining index has been shown to be directly proportional the aggressiveness of the clinical course (higher staining index, more aggressive).
References
WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. SH Swerdlow,et al.International Agency for Research on Cancer. Lyon, 2008. p. 229-231.
Robbins and Cotran Pathologic Basis of Disease. V Kumar, et al. 9th Edition. Elsevier Saunders. 2015. pp. 602-603.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127: 2375–2390. doi:10.1182/blood-2016-01-643569
Katzenberger T, Petzoldt C, Höller S, Mäder U, Kalla J, Adam P, et al. The Ki67 proliferation index is a quantitative indicator of clinical risk in mantle cell lymphoma. Blood. 2006;107: 3407. doi:10.1182/blood-2005-10-4079
Gao J, Peterson L, Nelson B, Goolsby C, Chen Y-H. Immunophenotypic variations in mantle cell lymphoma. Am J Clin Pathol. 2009;132: 699–706. doi:10.1309/AJCPV8LN5ENMZOVY
Boyd SD, Natkunam Y, Allen JR, Warnke RA. Selective immunophenotyping for diagnosis of B-cell neoplasms: immunohistochemistry and flow cytometry strategies and results. Appl Immunohistochem Mol Morphol. 2013;21: 116–131. doi:10.1097/PAI.0b013e31825d550a
Young KH, Chan WC, Fu K, Iqbal J, Sanger WG, Ratashak A, et al. Mantle cell lymphoma with plasma cell differentiation. Am J Surg Pathol. 2006;30: 954–961.
Gualco G, Weiss LM, Harrington WJ, Bacchi CE. BCL6, MUM1, and CD10 expression in mantle cell lymphoma. Appl Immunohistochem Mol Morphol. 2010;18: 103–108. doi:10.1097/PAI.0b013e3181bb9edf
Wang H-Y, Zu Y. Diagnostic Algorithm of Common Mature B-Cell Lymphomas by Immunohistochemistry. Arch Pathol Lab Med. 2017;141: 1236–1246. doi:10.5858/arpa.2016-0521-RA