- Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL)
- B-Cell Prolymphocytic Leukemia
- Spenic Marginal Zone Lymphoma (SMZL)
- Hairy Cell Leukemia (HCL)
- Splenic B-Cell Lymphoma/Leukemia, Unclassifiable
- Splenic Diffuse Red Pulp Small B-Cell Lymphoma
- Hairy Cell Leukemia-Variant
- Lymphoplasmacytic Lymphoma (LPL)
- Heavy Chain Diseases
- Plasma Cell Neoplasms
- Extranodal Marginal Zone Lymphoma of Mucosal Associated Lymphoid Tissue (MALT Lymphoma)
- Nodal Marginal Zone Lymphoma
- Follicular Lymphoma (FL)
- Primary Cutaneous Follicle Cell Lymphoma
- Mantle Cell Lymphoma
- Diffuse Large B-Cell Lymphoma (DLBCL), NOS
- T-Cell/Histiocyte-Rich Large B-Cell Lymphoma
- Primary DLBCL of the CNS
- Primary Cutaneous DLBCL, Leg Type
- EBV Positive DLBCL of the Elderly
- DLBCL Associated with Chronic Inflammation
- Lymphomatoid Granulomatosis (LYG)
- Primary Mediastinal (Thymic) Large B-Cell Lymphoma
- Intravascular Large B-Cell Lymphoma
- ALK Positive Large B-Cell Lymphoma
- Plasmablastic Lymphoma
- Large B-Cell Lymphoma Arising in HHV-8 Associated Multicentric Castleman Disease
- Primary Effusion Lymphoma
- Burkitt Lymphoma
- B-Cell Lymphoma, Unclassifiable (Indtermediate between DLBCL and Burkitt Lymphoma)
- B-Cell Lymphoma, Unclassifiable (Indtermediate between DLBCL and Hodgkin Lymphoma)
Category Archives: Organ Systems
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL)
CLL/SLL represents a B-cell neoplasm of small lymphocytes which involve a combination of peripheral blood, bone marrow, and/or lymph nodes. When peripheral blood predominates, then it is referred to as CLL, and when it presents as predominately nodal involvement it is referred to as SLL. This is the same disorder with different manifestations.
Continue reading Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL)
Monoclonal B-Cell Lymphocytosis (MBL)
Monoclonal B-cell lymphocytosis (MBL) was defined by the International Familial CLL consortium in 2005 as a monoclonal B-cell lymphocyte population in the peripheral blood <5,000/uL without evidence of lymphadenopathy (i.e. SLL), an autoimmune/infectious disease or other features diagnostic of a B-cell lymphoproliferative disorder. The 2016 WHO hematopathology revision dropped the requirement of cytopenias or disease related symptoms as adequate to make the diagnosis of CLL.
Follicular Lymphoma
Follicular Lymphoma (FL) is a mature B-cell lymphoma, which recapitulates or resembles germinal center B-cells. Most cases (~85%) harbor the characteristic t(14;18), which juxtaposes the BCL-2 gene on chromosome 18 with the IgH gene on chromosome 14 (and hence BCL-2 IHC protein expression). Most patients (~80-85) will present with advanced disease (stage III/IV), and bone marrow involvement is found in ~40% of cases with characteristic paratrabecular aggregates (mantle cell lymphoma and lymphoplasmacytic lymphoma may also have paratrabecular lymphoid aggregates). Most of the cases that lack the t(14;18) IgH/BCL-2 translocation (and are BCL-2 negative) are typically grade 3 FLs with a BCL-6 translocation (~10-15%). BCL-6 translocations can be evaluated for by FISH analysis, but the finding is NOT specific for FL.
Over time 30-50% of cases transform to diffuse large B-cell lymphoma (DLBCL). In a small subset of transformations, a second “hit” with a MYC translocation will occur resulting in a very aggressive high grade large B-cell lymphoma: the so-called “double hit” lymphoma.
Morphology
FL usually has at least a component of nodularity (+/- diffuse areas). There are two cell types that make up FL, centrocytes and centroblasts. Centrocytes are small cleaved cells with folded irregular nuclei. Centorblasts are large cells with more open chromatin, multiple nucleoli, and more cytoplasm compared to centrocytes.
Sometimes FL can have patterns that resemble marginal zone lymphoma, and can even have plasmacytic differentiation. Therefore, it is important that a panel of markers be used to identify (or exclude) evidence of germinal center differentiation. Occasional cases can have Hodgkin-like cells.
Immunophenotype
|
Marker
|
Comment
|
|
Negative
|
|
|
Positive
|
|
|
Positive
|
|
|
Positive
|
|
|
Positive (~90%), negative cases do not contain the t(14;18), which is more common in grade 3 cases
|
|
|
Positive, (~88%)
|
|
|
CD35
|
Highlights the follicular dendritic meshwork associated with FL.
|
|
Usually negative, higher grade lesions may be positive
|
|
|
Variable, shows low expression in low-grade processes, in distinct contrast to the high proliferation index and polarity associated with reactive germinal centers.
|
|
|
Negative
|
|
|
|
FL is typically expresses CD19, CD20, CD10, Bcl-6, and BCL-2 (~90%). CD5 is not expressed in FL.
- Normal reactive germinal centers do not express Bcl-2. In 90% of cases of FL, bcl-2 is expressed, which serves as a diagnostic tissue marker in lymphoma sections.
- CD23 expression by flow cytometry has been associated with lower grade FLs (e.g. grade 1 & 2) and better survival.
Grading
- Grade 1 & 2: <= 15 centroblasts/HPF (based on 0.159 mm² HPF)
- Grade 3: > 15 centroblasts/HPF (based on 0.159 mm² HPF)
- 3A: Centrocytes present in the background
- 3B: NO centrocytes present in the background (not associated with the IgH/BCL-2 rearrangement, and usually lacks expression of CD10 and BCL-2; often MUM-1+)
Grade 1 & 2 behave in a similar fashion as a low grade lymphoma. Grade 3 FL behaves as an intermediate grade lymphoma. Grading of FL with counting of large cells must take into consideration the field diameter of the microscope being used. The counts above are based on a F.N. 18 (0.159 mm² @ 40X). Most convention pathology scopes today are F.N. 22 (0.247 mm² @ 40X), and adjustments are necessary.
Pattern
- Predominately follicular: >75% follicular/nodular architecture
- Follicular and diffuse: 25-75% Diffuse areas or follicular/nodular architecture
- Preominately diffuse: <25% follicular/nodular areas (diffuse areas of otherwise grade 3 FL, then that component should be described as a separate component of diffuse large B-cell lymphoma)
Special Subtypes
- Large B-Cell Lymphoma with IRF4 Rearrangement
- Pediatric Follicular Lymphoma
- Occurs in children and young adults with an excellent prognosis, marked male predilection
- The morphology is high-grade (FL grade 3) appearing
- BCL-2 negative, lacK t(14;18)
- CD10 + (usually)
- MUM-1 negative
- Associated with TNFRSF14 deletions of mutations
- Localized process, usually in the head and neck area
- Duodenal Follicular Lymphoma
- Localized lesion
- Grade 1-2 pattern
- CD10/BCL-2 +
- t(14;18) present
- Lacks follicular dendritic meshwork
- Ki-67, low expression
- Excellent prognosis
- Predominately Diffuse Follicular Lymphoma with 1p36 deletion
- Localized mass (often inguinal)
- Diffuse pattern, grade 1/2
- Excellent prognosis
- Immunophenotype: CD20+, CD10+, BCL-2+, BCL-6+, CD23+ (subset of cases)
- t(14;18) NOT present
- 1p36 deletion (not specific)
- Lacks Bcl-2 rearrangement
- Primary Cutaneous Follicular Lymphoma
- In Situ Follicular Neoplasm (ISFN)
References
Robbins and Cotran Pathologic Basis of Disease. V Kumar, et al. 9th Edition. Elsevier Saunders. 2015. pp. 594-595.
Fedoriw Y, Dogan A. The Expanding Spectrum of Follicular Lymphoma. Surg Pathol Clin. 2016;9: 29–40. doi:10.1016/j.path.2015.11.001
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127: 2375–2390. doi:10.1182/blood-2016-01-643569
Xerri L, Dirnhofer S, Quintanilla-Martinez L, Sander B, Chan JKC, Campo E, et al. The heterogeneity of follicular lymphomas: from early development to transformation. Virchows Arch. 2016;468: 127–139. doi:10.1007/s00428-015-1864-y
MD DY-PW, BacSc F. A case of t (14; 18)-negative follicular lymphoma with atypical immunophenotype: usefulness of immunoarchitecture of Ki67, CD79a and follicular dendritic cell …. … Malaysian journal of …. 2014.
Boyd SD, Natkunam Y, Allen JR, Warnke RA. Selective immunophenotyping for diagnosis of B-cell neoplasms: immunohistochemistry and flow cytometry strategies and results. Appl Immunohistochem Mol Morphol. 2013;21: 116–131. doi:10.1097/PAI.0b013e31825d550a
Cook JR. Nodal and leukemic small B-cell neoplasms. Mod Pathol. 2013;26 Suppl 1: S15–28. doi:10.1038/modpathol.2012.180
Olteanu H, Fenske TS, Harrington AM, Szabo A, He P, Kroft SH. CD23 Expression in Follicular Lymphoma: Clinicopathologic Correlations. Am J Clin Pathol. 2011;135: 46–53. doi:10.1309/AJCP27YWLIQRAJPW
Gradowski JF, Jaffe ES, Warnke RA, Pittaluga S, Surti U, Gole LA, et al. Follicular lymphomas with plasmacytic differentiation include two subtypes. Mod Pathol. 2010;23: 71–79. doi:10.1038/modpathol.2009.146
Katzenberger T, Kalla J, Leich E, Stöcklein H, Hartmann E, Barnickel S, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113: 1053–1061. doi:10.1182/blood-2008-07-168682
Bayerl MG, Bentley G, Bellan C, Leoncini L, Ehmann WC, Palutke M. Lacunar and reed-sternberg-like cells in follicular lymphomas are clonally related to the centrocytic and centroblastic cells as demonstrated by laser capture microdissection. Am J Clin Pathol. 2004;122: 858–864. doi:10.1309/PMR8-6PHK-K4J3-RUH3
Lymphomatoid Granulomatosis
Lymphomatoid Granulomatosis (LYG) is a B-cell neoplasm of EBV + B-cells with characteristic anglocentric and angiodestructive features typically associated with some form of immunodeficiency.
LYG is an uncommon lymphoproliferative disorder and is not usually found in lymph nodes or spleen. Pulmonary involvement is present in >90% of cases, and other organs (e.g. brain, kidney, skin, liver) can be variably affected.
Morphology
LYG can have a varied appearance, but usually has a predominate background of small lymphocytes (T-cells), plasma cells, and histiocytes. Eosinophils and neutrophils are not a typical finding. Neoplastic EBV + B-cells can have a variable appearance ranging from immunoblast-like cells to morphologies similar to Hodgkin cells. The lymphoid infiltrate has an anglocentric and angiodestructive pattern, which may result in necrosis. If the pattern is of large cells in diffuse sheets, then the diagnosis of EBV+ diffuse large B-cell lymphoma should be made.
Differential diagnosis – Extranodal NK/T-cell lymphoma, nasal type is also an EBV+ tumor with an angiodestructive growth pattern.
Immunophenotype
Grading
- Grade 1 – < 5 EBV+ cells/HPF, polymorphic lymphoid infiltrate (large atypical cells rare/absent), No/focal necrosis.
- Grade 2 – 5-20 EBV+ cells/HPF (small clusters of B-cells by CD20), occasional large atypical/transformed cells.
- Grade 3 – > 50 EBV+ cells/HPF, numerous large atypical CD20+ B-cells (may form large aggregates)
References
WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. SH Swerdlow, et al. International Agency for Research on Cancer. Lyon, 2008. p. 247-249
Hairy Cell Leukemia
Hairy cell leukemia (HCL) is an uncommon (2% of lymphomas) but distinctive B-cell lymphoproliferative disease. Clinically, patients present with hepatosplenomegaly and pancytopenia usually with a marked moncytopenia. Classically, hairy cell leukemia has been difficult to differentiate from hairy cell variant (HCL-v) and splenic marginal zone lymphoma (SMZL). There has been significant morphologic, clinical and immunophenotypic overlap between these entities. An accurate diagnosis is critical as HCL is treated very differently (purine analogs) than other lymphomas with a high cure rate (>90%). Purine analog chemotherapy regimen is also much easier to tolerate than other chemotherapy protocols.
Morphology
HCL is characterized by small lymphoid cells with moderate to abundant cytoplasm (cells may be mistaken for erythroid precursors or other cells in the bone marrow trephine biopsy), which have an interstitial marrow pattern of involvement (morphologic estimate of involvement is often markedly underestimated compared to CD20 staining). Reticulin fibrosis is also characteristic, which often results in a “dry tap” (unable to obtain a bone marrow aspirate). Peripheral blood can show variable invovelemnt with characteristic “hair-like” cytoplasmic projections. It is very uncommon to have lymph node involvement.
Immunophenotype
- CD19/CD20 +
- CD25/CD103 + (CD25 co-expression with CD103 appears to be specific for HCL relative to the differential diagnosis with HCL-v and SMZL)
- Annexin A1 +
- CD11c +
- TRAP +
- DBA.44 + (thought to be specific for HCL when combined with TRAP +)
- CD5 – (rare cases <5% may be CD5+ by flow cytometry)
- CD10 -/+ (approximately 30% of cases may express CD10 by flow cytomtetry)
- BRAF VE1 (IHC) +
Molecular
- >90% found to have a BRAF V600E mutation. This can be identified by PCR or immunohistochemistry (IHC may be more sensitive than PCR). This appears to be relatively specific (relative to the usual differential diagnosis of HCL) with only rare cases of CLL/SLL, marginal zone lymphoma, and multiple myeloma found to express BRAF V600E IHC stain.
- MAP2K1 mutations (encodes MEK1 downstream of BRAF) in most BRAF negative HCL cases that use IgHV4-34. This mutation finding is not specific to HCL and may be seen in hairy cell variant (HCL-v).
References
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127: 2375–2390. doi:10.1182/blood-2016-01-643569
Uppal G, Ly V, Wang ZX, Bajaj R, Solomides CC, Banks PM, et al. The Utility of BRAF V600E Mutation-Specific Antibody VE1 for the Diagnosis of Hairy Cell Leukemia. Am J Clin Pathol. 2014;143: 120–125. doi:10.1309/AJCPQLQ89VXTVWKN
Brown NA, Betz BL, Weigelin HC, Elenitoba-Johnson KSJ, Lim MS, Bailey NG. Evaluation of Allele-Specific PCR and Immunohistochemistry for the Detection of BRAF V600E Mutations in Hairy Cell Leukemia. Am J Clin Pathol. 2014;143: 89–99. doi:10.1309/AJCPDN4Q1JTFGCFC
Boyd SD, Natkunam Y, Allen JR, Warnke RA. Selective immunophenotyping for diagnosis of B-cell neoplasms: immunohistochemistry and flow cytometry strategies and results. Appl Immunohistochem Mol Morphol. 2013;21: 116–131. doi:10.1097/PAI.0b013e31825d550a
Robbins and Cotran Pathologic Basis of Disease. V Kumar, et al. 9th Edition. Elsevier Saunders. 2015. pp. 603-604.
Turakhia S, Lanigan C, Hamadeh F, Swerdlow SH, Tubbs RR, Cook JR. Immunohistochemistry for BRAF V600E in the Differential Diagnosis of Hairy Cell Leukemia vs Other Splenic B-Cell Lymphomas. Am J Clin Pathol. 2015;144: 87–93. doi:10.1309/AJCP5WVXJ2KTLODO
Andrulis M, Penzel R, Weichert W, Deimling von A, Capper D. Application of a BRAF V600E mutation-specific antibody for the diagnosis of hairy cell leukemia. Am J Surg Pathol. 2012;36: 1796–1800. doi:10.1097/PAS.0b013e3182549b50
Mantle cell lymphoma
Mantle cell lymphoma (MCL) is a mature B-cell neoplasm, which represents 2-3% of cases of non-Hodgkin lymphoma in the US. The IgH/Cyclin D1 translocation is characteristic of this lymphoma, and can be confirmed by FISH testing in almost all cases. Clincially, patients often present with widespread disease. In addition to adenopathy, patient often have involvement of blood (20-40%) and other organ sites (gastrointestinal tract, liver spleen, bone marrow).
Morphology
MCL is typically characterized by a small to intermediate sized lymphocytes with an irregular nuclear membrane (CLL/SLL tends to have a smoother nuclear membrane and follicular lymphoma has cleaved cells). A subset of cases have a larger size and increased mitotic rate and can be confused with acute lymphoblastic lymphoma, and are referred to as the “blastoid” variant of MCL.
2008 WHO Classification identifies multiple morphologic variant including: blastoid variant (resembles ALL with increased mitoses), pleomorphic (also aggressive), small cell variant (resembles CLL/SLL), and marginal zone-like morphology. Architectural pattern can have a diffuse pattern (often with scattered histiocytes within the infiltrate) or nodular (resembling follicular lymphoma). Minimal lymph node involvement can show expansion of the mantle zones with relative intact lymph node architecture. Bone marrow involvement can have a varied appearance, but can mimic follicular lymphoma with paratrabecular aggregates.
2016 WHO Classification Revision
Two subtypes of mantle cell lymphoma are now recognized, which center around the mutation status of IgHV.
- IgHV unmutated/minimally mutated (mostly SOX11+) – classical disease that is aggressive, typically involves lymph nodes and other extra nodal sites.
- IgHV mutated (SOX11 negative) – associated with indolent non-nodal disease with peripheral blood and bone marrow involvement. Some of these cases may have been difficult to separate from CLL/SLL in the past.
Half of cyclin D1 negative cases show a CCND2 rearrangement.
Molecular Characteristics
- FISH + for t(11;14)
- Cytogenetics + t(11;14) ~70% of cases
- 50% of Cyclin D1 negative cases have CCND2 rearrangements
Immunophenotypic Expression Pattern
|
Marker
|
Comment
|
|
Negative
|
|
|
Positive (93-95%). Some data indicates up to 12% of MCL cases may be negative for CD5.
|
|
|
Negative. Up to 8% of cases may express CD10 (expression will usually be <30%).
|
|
|
Positive
|
|
|
Positive
|
|
|
Negative. 21% may express CD23.
|
|
|
Positive. Nuclear expression. The rabbit monoclonal antibody clone SP4 appears to have the highest sensitivity and stain intensity. Sensitivity ~95%.
|
|
|
Positive
|
|
|
Negative (~12% of cases may have expression)
|
|
|
Usually negative (35% may be positive, of these 2/3rds will also be bcl-6+)
|
|
|
Highlights the residual FDC meshwork.
|
|
|
Inverse relationship between quantitative Ki-67 index and prognosis. Ki-67 >40% is an adverse prognostic factor.
|
|
| Expressed in classic form and lack of expression is associated with more indolent variant of MCL. |
Important caveats
Cyclin D1 expression is not entirely specific for mantle cell lymphoma. Some expression can be seen in a zonal pattern in CLL/SLL in proliferation centers. Other entities such as plasma cell myeloma (up to 50%) and hairy cell leukemia can also express cyclin D1. CD10 (8%), Bcl-6, and CD23 (21%) has been reported to be expressed in a small number of cases. Loss of CD5 (12%) has been noted in some cases. Most of these studies were by flow cytometry.
CD5 expression on a B-cell lymphoma should practically result in testing for exclusion of a t(11;14) IgH/Cyclin D1 gene fusion. There are other B-cell lymphomas, which may also have CD5 expression (e.g. DLBCL, CLL/SLL, etc.) that are not MCL and have separate prognostic characteristics because of/or separate from CD5 expression.
Rarely, plasmacytic differentiation may be identified. Classic MCL is derived form naive B-cells, which tends to not to progress to plasmacytic differentiation, compared to post germinal center derived B-cell lesions, which more commonly have plasmacytic differentiation.
Ki-67 staining index has been shown to be directly proportional the aggressiveness of the clinical course (higher staining index, more aggressive).
References
WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. SH Swerdlow,et al.International Agency for Research on Cancer. Lyon, 2008. p. 229-231.
Robbins and Cotran Pathologic Basis of Disease. V Kumar, et al. 9th Edition. Elsevier Saunders. 2015. pp. 602-603.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127: 2375–2390. doi:10.1182/blood-2016-01-643569
Katzenberger T, Petzoldt C, Höller S, Mäder U, Kalla J, Adam P, et al. The Ki67 proliferation index is a quantitative indicator of clinical risk in mantle cell lymphoma. Blood. 2006;107: 3407. doi:10.1182/blood-2005-10-4079
Gao J, Peterson L, Nelson B, Goolsby C, Chen Y-H. Immunophenotypic variations in mantle cell lymphoma. Am J Clin Pathol. 2009;132: 699–706. doi:10.1309/AJCPV8LN5ENMZOVY
Boyd SD, Natkunam Y, Allen JR, Warnke RA. Selective immunophenotyping for diagnosis of B-cell neoplasms: immunohistochemistry and flow cytometry strategies and results. Appl Immunohistochem Mol Morphol. 2013;21: 116–131. doi:10.1097/PAI.0b013e31825d550a
Young KH, Chan WC, Fu K, Iqbal J, Sanger WG, Ratashak A, et al. Mantle cell lymphoma with plasma cell differentiation. Am J Surg Pathol. 2006;30: 954–961.
Gualco G, Weiss LM, Harrington WJ, Bacchi CE. BCL6, MUM1, and CD10 expression in mantle cell lymphoma. Appl Immunohistochem Mol Morphol. 2010;18: 103–108. doi:10.1097/PAI.0b013e3181bb9edf
Wang H-Y, Zu Y. Diagnostic Algorithm of Common Mature B-Cell Lymphomas by Immunohistochemistry. Arch Pathol Lab Med. 2017;141: 1236–1246. doi:10.5858/arpa.2016-0521-RA
Lymphoplasmacytic Lymphoma (LPL)
Lymphoplasmacytic Lymphoma (LPL)
- Neoplastic proliferation of B-cells ranging in spectrum from lymphocytes to plasma cells (typically involves the bone marrow, but can also involve the spleen and lymph nodes).
- Bone marrow involvement
- Nodular, diffuse, and/or interstitial
- Small lymphocytes admixed with plasma cells and plasmacytoid lymphocytes
- IgM paraprotein
- Sometimes result in a hyperviscosity syndrome(30%)
- IgM and IgG paraprotein (minority)
- Some cases may be IgG or IgA.
- Cryoglobulinemia (20% of WM)
- Coagulopathy (IgM binds to clotting factors)
- MYD88 (L265P) point mutation (>90%)
- Results in up-regulation of NF-κB (promotes tumor cell survival).
- Not specific for LPL.
- LPL is a diagnosis of exclusion after other B cell lymphoid neoplasms with plasmacytic differentiation have been excluded.
- IgM MGUS is now thought to be more closely related to LPL than plasma cell myeloma.
Most patients present with non-specific B-symptoms. However, ~10% have hemolysis secondary to cold agglutinins (IgM binds to RBCs at temperature <37C).
Waldenstrom macroglobulinemia
Waldenstrom macroglobulinemia (defined as bone marrow involvement by LPL and IgM monoclonal gammopathy) may manifest as a hyperviscosity syndrome due to the circulating IgM in the blood (IgM tends to stay in the vascular system, compared to IgG which can penetrate into soft tissue). Because IgM molecules tend to be well-segregated to the vascular compartment, plasmapheresis can be very effective to treat hyerpviscosity symptoms. Clinical signs/symptoms include:
- Cryoglobulinemia – precipitation of macroglobulins as temperatures <37C
- Bleeding – macroglobulins interfere with clotting factors
- Neurologic disturbances – secondary to increased blood viscosity
- Visual impairment – secondary to increased viscosity and hemorrhage
Immunophenotype
|
Marker
|
Comment
|
|
Negative
|
|
|
Negative (~10% +)
|
|
|
Negative
|
|
|
Positive
|
|
|
Negative (rare atypical cases +)
|
|
|
Positive (best marker). Marks from Mature B-Cells through Plasma Cell differentiation.
|
|
|
Often Positive
|
|
|
Positive
|
|
|
Negative
|
|
|
Positive
|
Morphology
The bone marrow can have a varying degree of involvement (usually interstitial to nodular) by tumor cells showing a range from small lymphocytes to plasmacytoid lymphocytes to plasma cells. Mast cell hyperplasia is commonly present.
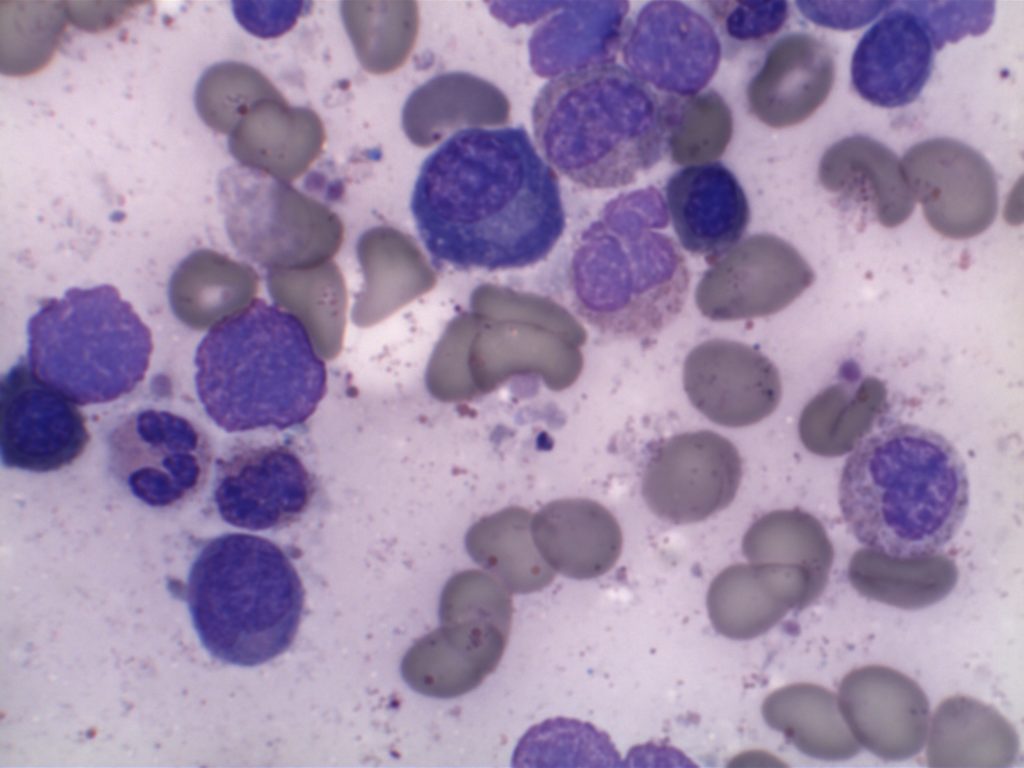
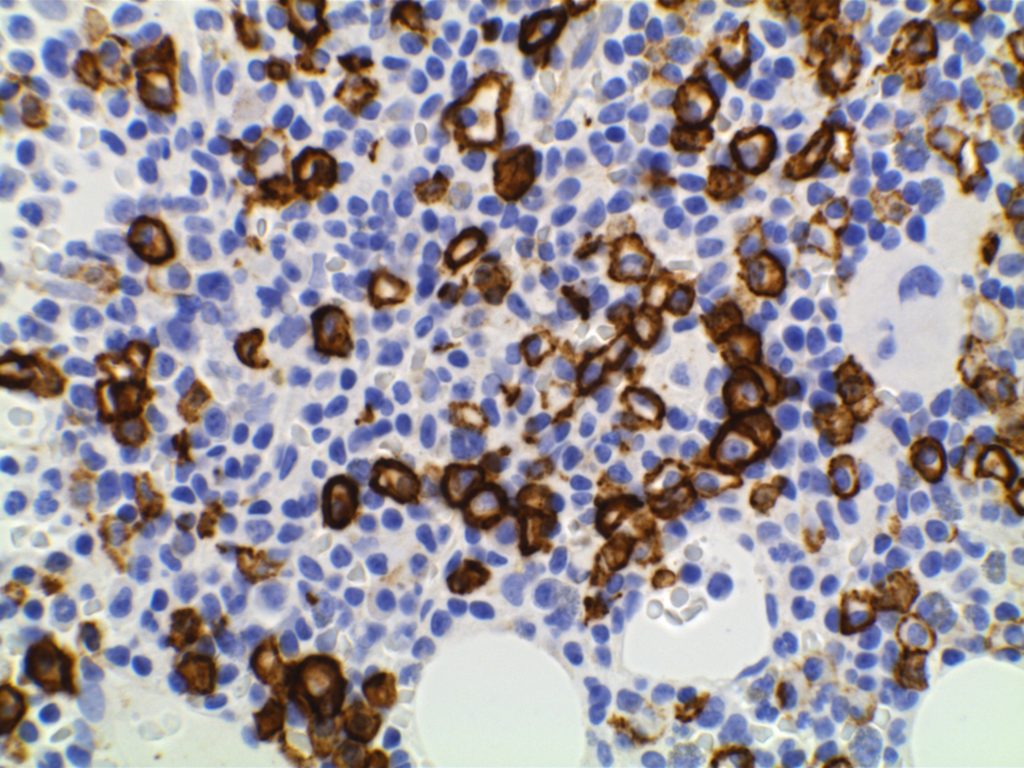
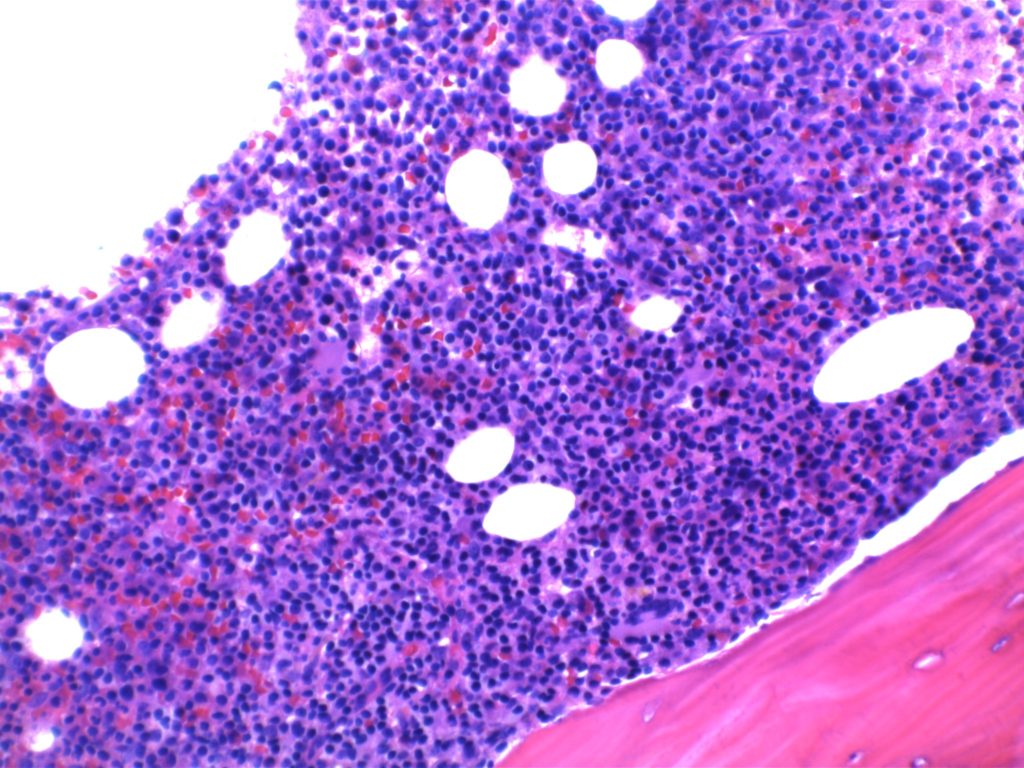
References
Robbins and Cotran Pathologic Basis of Disease. V Kumar, et al. 9th Edition. Elsevier Saunders. 2015. pp. 601-602.
Harmon CM, Smith LB. B-cell Non-Hodgkin Lymphomas with Plasmacytic Differentiation. Surg Pathol Clin. 2016;9: 11–28. doi:10.1016/j.path.2015.09.007
WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. SH Swerdlow, et al. International Agency for Research on Cancer. Lyon, 2008. p. 194-195
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127: 2375–2390. doi:10.1182/blood-2016-01-643569
Martinez-Lopez A, Curiel-Olmo S, Mollejo M, Cereceda L, Martinez N, Montes-Moreno S, et al. MYD88 (L265P) somatic mutation in marginal zone B-cell lymphoma. Am J Surg Pathol. 2015;39: 644–651. doi:10.1097/PAS.0000000000000411
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367: 826–833. doi:10.1056/NEJMoa1200710
Bone Marrow IHC. Torlakovic, EE, et. al. American Society for Clinical Pathology Pathology Press © 2009. pp. 27.
Hematopathology. [edited by] Jaffe, ES. 1st. ed. Elsevier, Inc. © 2011. pp.194-195.
MYD88 (L265P) Mutation
MYD88 (L265P) mutation has garnered a lot of excitement because of its usefulness in diagnosing lymphoplasmacytic lymphoma (LPL). Traditionally, LPL has been difficult to diagnose because exclusion of other B-cell neoplasms with plasmacytic differentiation is required. MYD88 mutations are present in approximately 90% of cases of LPL, and in a much lower percentage of cases of B cell lymphomas, which are typically in the differential diagnosis of LPL.
MYD88 mutation frequency
- LPL ~ 90%
- DLBCL ~ 30% (non-germinal center)
- DLBCL >50% (primary cutaneous, leg type)
- Multiple myeloma – negative (even IgM subtype)
- CLL/SLL – 2-3%
- Extranodal marginal zone lymphoma – 7%
- Splenic marginal zone lymphoma – 4-15%
- Nodal marginal zone lymphoma – rare
References
Harmon CM, Smith LB. B-cell Non-Hodgkin Lymphomas with Plasmacytic Differentiation. Surg Pathol Clin. 2016;9: 11–28. doi:10.1016/j.path.2015.09.007
WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. SH Swerdlow, et al. International Agency for Research on Cancer. Lyon, 2008. p. 194-195
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127: 2375–2390. doi:10.1182/blood-2016-01-643569
Martinez-Lopez A, Curiel-Olmo S, Mollejo M, Cereceda L, Martinez N, Montes-Moreno S, et al. MYD88 (L265P) somatic mutation in marginal zone B-cell lymphoma. Am J Surg Pathol. 2015;39: 644–651. doi:10.1097/PAS.0000000000000411
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367: 826–833. doi:10.1056/NEJMoa1200710
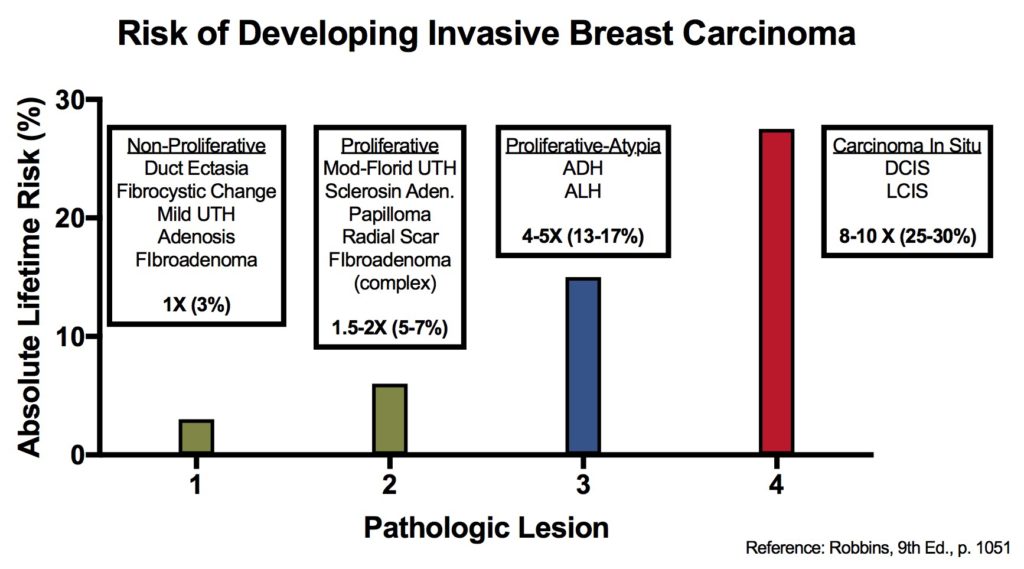
Breast – Atypical Lobular Hyperplasia (ALH)
Atypical lobular hyperplasia is a proliferation of epithelial cells in the terminal duct-lobular unit that lacks expression of E-cadherin and fills the lobular unit (but does not expand). Some define ALH as filling or distending <50% of the acini within an affected lobule. There is some variation between experts as the exact differentiating line between ALH and lobular carcinoma in situ (LCIS). It is best to think of ALH and LCIS as representing a morphologic spectrum with filling and distention of lobular acini being the measured characteristic.
In some cases ALH/LCIS may appear similar to DCIS or merge with areas of DCIS. E-cadherin is a helpful marker to differentiate between these lesions. Clinically it is important to differentiate ALH/LCIS from DCIS because LCIS/ALH is an incidental finding on mammogram and represents a risk factor for bilateral breast carcinomas. Management is not through local excision, but surveillance and hormonal therapy dependent upon the type of lesion. DCIS is treated through local excision because these lesions tend to be localized/focal.
ALH carries an increased risk of developing an invasive breast carcinoma (4-5x relative risk, 13-17% lifetime risk), while LCIS carries an 8-10x increased relative risk, 25-30% lifetime risk). These are the same risks as ALH and DCIS, respectively. Although ALH/LCIS risk is bilateral, while ADH/DCIS tends to be more ipsilateral.
Breast lesions and risk of developing an invasive carcinoma
References
Kumar, Vinay, Abul K. Abbas, and Jon C. Aster. Robbins and Cotran Pathologic Basis of Disease. Ninth edition. Philadelphia, PA: Elsevier/Saunders, 2015. p. 1050-1051